
Discussion
Lysinuric Protein Intolerance (LPI) and Pulmonary alveolar proteinosis (PAP)
Lysinuric protein intolerance (LPI) is an autosomal recessive transport disorder of the cationic amino acids lysine, arginine, and ornithine [1].
It is caused by mutations in the SLC7A7 (solute carrier family 7, member 7) gene encoding the y+L amino acid transporter y+LAT-1) [1].
LPI is more prevalent in Finland (1 in 60 000) than elsewhere in the world [1].In LPI, the transport defect is located at the basolateral cell membrane of polar epithelial cells in the intestine and renal tubules [1]. Urinary excretion of lysine, arginine, and ornithine is massively increased and their absorption from the intestine is decreased. The deficiency of urea cycle intermediates arginine and ornithine causes urea cycle dysfunction and episodes of hyperammonemia after ingestion of protein.
The characteristic symptoms and signs of LPI include:
- aversion to dietary protein
- postprandial vomiting
- failure to thrive
- growth retardation
- osteoporosis
- hepatosplenomegaly
- muscle weakness.
Mental development is normal if hyperammonemia can be avoided. Several patients with LPI have developed pulmonary alveolar proteinosis and renal insufficiency [1]. In patients with LPI, birth weights and lengths are usually normal for gestational age, and postnatal growth is normal for as long as the infants are breast-fed. After weaning, the growth curves begin to deviate progressively from the normal mean and the skeletal maturation is delayed 1] Osteopenia and a pronounced tendency to fractures are constant complications of LPI, and osteopenia may even be the presenting sign of LPI [1]. Most patients have structural abnormalities in the skeleton, but there is no correlation between fracture incidence and radiologic bone structure [1].
The patients do not usually show clinical nor biochemical signs of rickets. Osteopenia in LPI may thus reflect defective extracellular matrix protein synthesis due to dietary protein deprivation and functional deficiency of cationic or other essential amino acids [1]. The treatment of LPI consists of dietary protein restriction and oral supplementation with citrulline, a neutral amino acid and urea cycle intermediate that is effectively absorbed in LPI [1]. It improves protein tolerance and helps to prevent hyperammonemia.
Of the 38 LPI patients diagnosed in Finland since 1965, four pediatric patients had died by 1994 and the clinical course has been reported. All patients developed acute respiratory insufficiency [2]. In addition to pulmonary hemorrhages, three of the patients had PAP and one had cholesterol granulomas. Three patients had a clinically obvious renal insufficiency, but all four showed histologic signs of immune complex mediated glomerulonephritis. The patients also developed hepatic insufficiency with fatty degeneration or cirrhosis. All patients showed anemia, thrombocytopenia, and a severe bleeding tendency. The bone marrow of three patients was hypercellular, but the amount of megakaryocytes was decreased in two cases. Amyloid was present in the lymph nodes and the spleen. Bone specimens showed osteoporosis. One additional pediatric LPI patient with fatal respiratory insufficiency was reported previously. In that case the predominant histologic pattern changed over time from endogenous lipoid pneumonia to PAP. The relationships between pulmonary diseases characterized by endogenous lipid accumulation have been questioned since their initial descriptions. Several studies have proven a connection between these diseases [3]. PAP and cholesterol granulomas, the two main histologic findings in the lung specimens of patients with LPI, could represent different stages of the same pulmonary reaction rather than separate diseases.
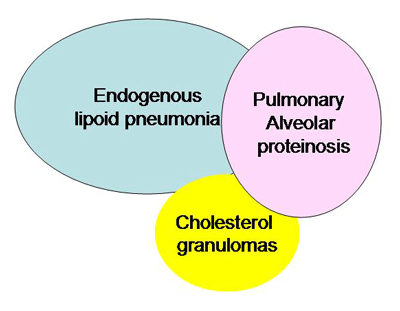
Figure 8
PAP is a rare cause of chronic interstitial lung disease in children, characterised by accumulation of a lipoproteinaceous material in the alveoli [4]. Bronchoalveolar lavage is the key diagnostic tool, revealing a milky opaque appearance of the return fluid and a periodic acid-Schiff staining material in the alveolar macrophages. PAP is a heterogeneous disease. Immediate-onset forms leading to early and fatal respiratory failure may be related to SP-B deficiency. Postnatal-onset PAP may be associated with various diseases or may be primary. The latter has a polymorphic progression from asymptomatic to uncontrollable respiratory failure. Recent studies have implicated GM-CSF and/or its receptor but the exact underlying mechanisms are still unknown. Therapeutic lung lavages have been considered the primary treatment but in younger children this may not be the as effective in severe cases as reported in the adult literature.
The underlying theme is of a phospholipid metabolism disorder and attention to this aspect may lead onto a unifying mechanism for these rare but troublesome diseases.
Summary
- Clinical and pathophysiological heterogeneity with PAP
- Whole lung lavage in the younger age group is doable but not always successful, particularly in the youngest.
- Outcome depends on the cause and is modified by additional co-morbidity.
- Rich area for applied scientific research